
Un essai clinique est une recherche pratiquée sur l’Homme pour évaluer l’efficacité et la tolérance d’une méthode diagnostique, d’un traitement, d’un dispositif médical.
Quand cela concerne le médicament, un essai clinique a pour objectif, par exemple, d’établir ou de vérifier les données sur le devenir du médicament dans l’organisme, ou bien son mécanisme d’action, ou son efficacité et sa tolérance.
Pour un dispositif médical, l’essai clinique peut avoir comme objectif la détermination de ses performances et la mise en évidence d’éventuels effets indésirables.
L’essai clinique peut se faire chez une personne malade ou une personne non malade (volontaire sain).
Les DM (dispositif médical), DMDIV (dispositif médical de diagnostic in vitro) et médicaments ont des définitions bien précises. Pour être commercialisés, les DM et les DMDIV doivent obtenir une certification européenne et les médicaments une autorisation des autorités de santé. Ce sont des produits qui doivent répondre à une règlementation spécifique.
Dispositif Médical (DM)
Un dispositif médical est un produit de santé utilisé chez l’homme pour une finalité diagnostique ou thérapeutique, pour compenser un handicap ou comme moyen de maîtrise de la conception. Ces produits de santé agissent selon un mécanisme d’action « mécanique », c’est-à-dire qu’il n’est ni pharmacologique (effet du produit sur l’organisme), ni immunologique (effet sur le système immunitaire), ni métabolique (effet sur le fonctionnement des cellules de l’organisme).
Les dispositifs médicaux se présentent sous plusieurs formes : implant, équipement, applications mobiles, etc. Pour être commercialisés, ils doivent obtenir une certification (marquage CE médical) qui garantit que ces dispositifs répondent à des exigences spécifiques de sécurité et de bénéfice clinique pour la personne.
Il existe plusieurs classes de dispositifs médicaux, en fonction d’un risque croissant :
Certains dispositifs médicaux peuvent incorporer un médicament. Si l’effet recherché est lié au médicament, le produit de santé est alors considéré comme un médicament et doit se conformer à la réglementation des médicaments.
Quelle que soit la classe du DM, les exigences de sécurité et de performance clinique sont les mêmes.
Pour en savoir plus : snitem.fr
Dispositif médical de diagnostic in vitro (DMDIV)
Un dispositif médical de diagnostic in vitro est un dispositif qui consiste en un réactif, un produit réactif, un matériau d’étalonnage, un matériau de contrôle, une trousse, un instrument, un appareil, un équipement, un logiciel ou un système utilisé seul ou en association, destiné à être utilisé in vitro dans l’examen d’échantillons provenant du corps humain (sang, selles, urine, cellules…), dans le but de fournir des informations :
Il existe 4 classes de DMDIV (classe A, B, C et D) en fonction de leur destination.
Ils sont utilisés la plupart du temps par les professionnels de santé (médecin, biologistes…)
Quelques exemples : autotests (tests de grossesse), tests génétiques, tests de dépistage de cancer, diagnostics compagnons, réactifs de dosage (glycémie, cholestérol…) …
Pour être commercialisés, ils doivent obtenir une certification (marquage CE) qui garantit que ces dispositifs répondent à des exigences spécifiques de sécurité et de performance.
Pour en savoir plus : sidiv.fr
Logiciels et applications mobiles
Les logiciels et les applications mobiles dans le domaine de la santé, sont considérés comme des dispositifs médicaux ou des dispositifs médicaux de diagnostics in vitro lorsqu’ils répondent aux critères suivants :
Les logiciels ou applications mobiles destinés à s’assurer de la bonne prise d’un traitement par un patient, à préparer l’entraînement de sportifs, à compter le nombre de pas… ne sont pas des dispositifs médicaux ou des dispositifs médicaux de diagnostic in vitro car ils ne répondent pas aux critères précédemment cités.
Médicament
On entend par médicament toute substance ou composition présentée comme possédant des propriétés curatives ou préventives à l’égard des maladies humaines ou animales, ainsi que toute substance ou composition pouvant être utilisée chez l’homme ou l’animal, en vue d’établir un diagnostic médical ou de restaurer, corriger ou modifier leurs fonctions physiologiques en exerçant une action pharmacologique, immunologique ou métabolique.
Le médicament peut être un médicament chimique, un médicament biologique (par exemple un vaccin) ou un médicament à base de plantes.
Pour être commercialisé, le médicament doit obtenir une Autorisation de mise sur le marché (AMM).
Pour en savoir plus : leem.org
Afin de savoir si un médicament ou un dispositif médical va être aussi bon ou même meilleur que ce qui est déjà disponible (notamment en termes d’efficacité et/ou de tolérance), il est indispensable de faire des comparaisons de ces différents traitements ou dispositifs par un essai clinique.
Plusieurs groupes de personnes sont ainsi traitées en parallèle par les médicaments ou les dispositifs médicaux que l’on souhaite comparer.
Essai contre placebo ou contre comparateur ?
Il y a deux sortes d’essais :
Rien n’empêche qu’une personne prenant le placebo ou le traitement évalué n’ait accès à d’autres traitements si son état de santé l’exige.
Si à tout moment au cours d’un essai l’investigateur a des raisons de craindre un désavantage réel pour une personne participant à l’essai, quel que soit le groupe de traitement (placebo ou traitement évalué), il peut décider de sortir de l’essai la personne concernée.
Dans ce cas, la personne pourra recevoir un nouveau traitement. Ce nouveau traitement est donné du fait de l’efficacité jugée insuffisante du traitement reçu ou d’effets indésirables auxquels il faut mettre fin.
Ceci n’enlève en rien au patient la possibilité de pouvoir sortir de l’essai à tout moment sans avoir à se justifier.
Toutes les recherches impliquant une personne doivent obtenir un avis favorable d’un comité de protection des personnes (CPP) avant leur mise en œuvre.
Toutes les recherches impliquant une personne doivent obtenir un avis favorable d’un comité de protection des personnes (CPP) avant leur mise en œuvre.
Le CPP rend un avis sur les conditions de validité de la recherche au regard notamment de la protection des patients, de l’information qui leur est donnée, de la pertinence de la recherche, des bénéfices et des risques attendus.
De plus, pour certaines recherches, il est indispensable que l’agence nationale de sécurité du médicament et des produits de santé (ANSM) se prononce également sur certains aspects scientifiques. C’est notamment le cas pour les recherches interventionnelles portant sur le médicament et les recherches portant sur des dispositifs médicaux à risque (implants par exemple) qui n’ont pas encore obtenu leur marquage CE.
L’ANSM se prononce au regard de la sécurité des personnes par rapport à la qualité, la sécurité et les conditions d’utilisation du produit à évaluer ainsi que des modalités de surveillance des personnes.
Toutes les recherches doivent également obtenir une autorisation de la CNIL (commission nationale de l’informatique et des libertés) qui s’assurera que la mise en œuvre des traitements informatiques des données garantit bien la protection des données personnelles des participants et le respect de leur vie privée.
Pour aller plus loin, lisez l’édito d’Alain Le Hénaff Les CPP : « Un regard croisé de différentes compétences et de diverses sensibilités »
Plusieurs mécanismes permettent d’assurer la sécurité des personnes pendant toute la durée d’un essai :
Ainsi, des mesures correctrices peuvent être prises à tout moment pour assurer la sécurité des personnes.
Avant le commencement d’un essai clinique, le promoteur doit avoir souscrit une assurance garantissant sa responsabilité civile et celle de tout intervenant dans l’essai afin de garantir les éventuels préjudices liés à la participation d’une personne à un essai.
Cette assurance est obligatoire pour certains essais, notamment ceux pendant lesquels la pratique médicale courante est modifiée.
🎧+ ℹ️ Sans recherche clinique, pas de progrès médical.
La recherche est indispensable au progrès des soins.
Ce progrès est possible par l’amélioration des connaissances scientifiques, par les recherches menées dans le but de mieux comprendre les mécanismes d’apparition et de développement des pathologies.
La médecine ne sait pas encore guérir certaines maladies, les chercheurs doivent trouver de nouvelles stratégies thérapeutiques (nouveaux médicaments, nouvelles associations de médicaments) pour mieux soigner les malades. Le progrès médical concerne également la prévention et le diagnostic des maladies, les techniques et le matériel utilisés pour les soins et la chirurgie, les outils pour aider à l’interprétation de données recueillies chez les patients.
Afin de proposer ces innovations à toutes les personnes qui peuvent en bénéficier, elles doivent être évaluées chez l’Homme par de la recherche clinique.
La recherche clinique couvre l’ensemble des recherches organisées et pratiquées sur l’Homme en vue du développement des connaissances biologiques et médicales.
Chaque essai clinique est conduit pour répondre à une question précise.
Pour aller plus loin : entretien avec la Prof. Martine Laville qui nous explique à quoi sert la recherche clinique
Au-delà de ces différents termes, il faut retenir la notion de Recherche Impliquant la Personne Humaine (RIPH). Il s’agit de recherches qui sont organisées et pratiquées sur des personnes malades ou non malades (appelées volontaires sains), en vue du développement des connaissances biologiques ou médicales.
Les recherches impliquant la personne humaine (RIPH) viseront à évaluer :
D’autres termes sont parfois utilisés : recherche clinique, étude clinique, essai clinique.
Lorsque la recherche impliquant la personne humaine a pour objectif de s’assurer de l’efficacité et/ou de la sécurité d’un médicament ou de comparer plusieurs médicaments entre eux, on parle souvent d’étude clinique ou d’essai clinique. Lorsque ces recherches concernent des dispositifs médicaux, on parle d’investigation clinique.
Dans le langage courant, les termes « recherche », « étude », « essai » et « investigation » sont employés indifféremment, on parle donc de la même chose : recherche qui implique la personne humaine.
L’accès précoce et l’accès compassionnel sont des dispositifs de mise à disposition des patients de médicaments ne possédant pas encore d’autorisation de mise sur le marché ou utilisés en dehors de leurs indications thérapeutiques validées, mais pour lesquels des données de sécurité et d’efficacité sont suffisantes pour permettre leur administration à un malade.
Ces dispositifs permettent un accès précoce aux traitements présumés innovants pour des patients en impasse thérapeutique et qui ne peuvent pas avoir accès à des essais cliniques. Les médicaments qui entrent dans ces dispositifs sont sous surveillance étroite, notamment en ce qui concerne la sécurité (signalement des effets secondaires).
| Accès précoce
|
Accès compassionnel
|
|
| Pour qui ? | Un groupe de malades traités et suivis selon des critères précis définis dans un protocole d’utilisation thérapeutique et de recueil d’information
|
Un malade nommément désigné et ne pouvant pas participer à une recherche clinique en cours avec le médicament. Le malade est suivi selon des critères définis dans un protocole d’utilisation thérapeutique |
| Qui fait la demande ? | Le titulaire des droits d’exploitation (en général une entreprise pharmaceutique)
|
Le médecin prescripteur |
| Pour quel médicament ? | Un médicament dont l’efficacité et la sécurité d’emploi sont fortement présumées au vu des données disponibles et dont la demande d’autorisation de mise sur le marché est en cours d’évaluation ou la demande de décision sur la prise en charge par l’Assurance Maladie est en attente
|
Un médicament dont le rapport efficacité/sécurité, au vu des données disponibles, est présumé favorable pour le malade et pour lequel aucune recherche clinique n’est en cours |
| Qui donne l’autorisation ? | La Haute Autorité de Santé (HAS), après avis favorable de l’ANSM
|
L’ANSM |
| Qui effectue la prise en charge ? | L’Assurance Maladie
|
L’Assurance Maladie |
Ces dispositifs sont différents d’une recherche clinique sur plusieurs points :
| Recherche clinique | Accès précoce ou compassionnel
|
|
| Objectif | Tester un nouveau médicament | Donner un accès à un nouveau traitement présumé innovant ou un médicament en dehors de ses indications habituelles, à des patients en échec de traitement
|
| Lieu de dispensation | Dans quelques centres investigateurs déterminés | Dans les établissements de santé dans le cadre d’une hospitalisation ou en rétrocession
|
| Critères d’octroi du médicament au patient | Critères d’inclusion et d’exclusion bien définis
|
Indications établies par l’ANSM et la HAS
|
| Qui demande l’autorisation ? | Le promoteur de la recherche
|
En général le médecin prescripteur
|
| A quel stade du développement d’un médicament ? | Tous
|
Lorsque les données de sécurité et d’efficacité disponibles sont présumées favorables, juste avant l’obtention de l’AMM ou lors de l’attente du remboursement
|
| Quel suivi des patients ? | Suivi pendant toute la durée de la recherche
|
Suivi du malade par le médecin prescripteur pendant la durée du traitement
|
| Qui finance ? | Le promoteur de la recherche
|
La Sécurité Sociale
|
Pour le dispositif médical et le dispositif médical de diagnostic in vitro, une prise en charge dérogatoire et temporaire est possible afin de favoriser l’accès rapide des patients à des technologies innovantes en phase précoce de développement clinique. Il s’agit du « forfait innovation ». L’obtention de ce forfait est conditionnée à la réalisation d’une étude visant à fournir des données cliniques ou médico- économiques manquantes. Les dispositifs concernés doivent avoir des caractères de nouveauté, de sécurité pour les patients et apporter un bénéfice clinique important permettant de satisfaire un besoin médical non couvert ou réduire de façon significative les dépenses de santé.
Par exemple, ce système a concerné un implant sous rétinien pour des patients souffrant de dégénérescence rétinienne, ou un système de traitement d’adénomes du sein par des ultrasons, et également un cœur artificiel.
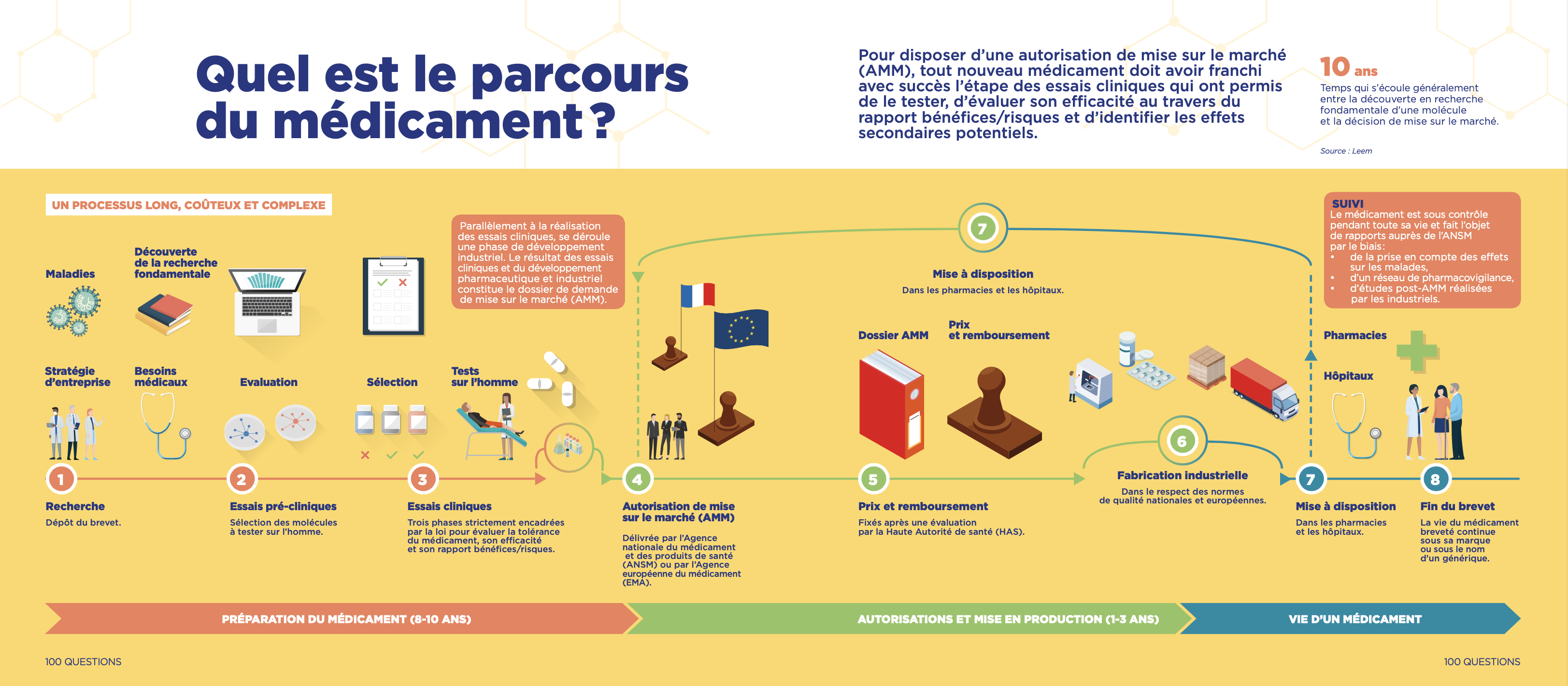
ℹ️ Pour un médicament, l’évaluation de son efficacité et de sa sécurité s’effectue en général en 4 phases.
Tous ces essais cliniques devront figurer dans le dossier préparé en vue d’obtenir une autorisation de mise sur le marché.
Pour aller plus loin :

Source : Leem
ℹ️ Pour un dispositif médical, l’évaluation de ses performances et de sa sécurité dans le but d’obtenir un marquage CE (conformité à la réglementation européenne) permettant sa mise sur le marché, impose une évaluation clinique.
Pour les dispositifs non implantables de faible et moyen degré de risque (par exemple, des fauteuils roulants, des scalpels, des lentilles de contact, des couronnes dentaires, des échographes…), cette évaluation clinique peut se faire en utilisant les données existantes comme des publications scientifiques. Pour les dispositifs implantables et les dispositifs de classe III (à degré de risque élevé, prothèses par exemple), un essai clinique est indispensable.
Une surveillance après commercialisation est également prévue.
Pour aller plus loin :


Source : Snitem
Les différents types de recherche clinique sur la personne sont définis dans des textes législatifs.
Ces recherches se différencient par :
En règle générale, on distingue :
Pour les dispositifs médicaux, une distinction se fait selon le marquage CE :
De nombreux intervenants participent au bon déroulement d’un essai clinique.
Les principaux acteurs sont :
Dans un essai clinique de comparaison entre un nouveau traitement (médicament ou dispositif médical) et un traitement déjà disponible, les personnes qui participent à l’essai sont traitées en même temps dans plusieurs groupes différents.
Chacun des groupes prend un des traitements que l’on souhaite comparer.
Après avoir vérifié que la personne répond aux critères d’éligibilité de l’essai et que son consentement a été recueilli, l’investigateur peut inclure la personne dans l’essai.
Pour garantir l’objectivité de l’investigateur, ce n’est pas lui qui choisit le groupe dans lequel la personne est incluse.
Le type de traitement qui sera reçu par la personne est déterminé par tirage au sort informatique : le terme technique utilisé est « randomisation ».
Le tirage au sort est le seul moyen de garantir une comparaison scientifiquement acceptable entre deux groupes de personnes traitées.
Le traitement qui sera ensuite donné à la personne sera appliqué et contrôlé par l’investigateur qui la suit.
Dans un essai ouvert, l’investigateur et le patient connaissent l’ensemble des traitements pris (médicament ou dispositif médical).
Les essais en ouvert peuvent être menés sur une plus longue période et permettre de donner plus d’information sur la sécurité d’un traitement.
Dans un essai en double aveugle, ni l’investigateur ni le personnel soignant ni le patient ne savent quel traitement est reçu (placebo, traitement de référence, traitement évalué).
La technique de l’essai en double aveugle est souvent utilisée pour éviter que l’investigateur, l’équipe soignante et le patient ne soient influencés par une opinion a priori qu’ils pourraient avoir sur tel ou tel traitement.
Il est difficile de mettre en place un essai en double aveugle pour un dispositif médical car il s’agit d’un produit utilisateur dépendant et dans la majorité des cas l’utilisateur (investigateur ou patient) sait ce qu’il utilise.
🎧 Les médicaments et les dispositifs médicaux, lorsqu’ils sont destinés à l’enfant ou à l’adolescent, ne peuvent être uniquement évalués chez l’adulte.
Il faut donc faire des essais chez l’enfant ou l’adolescent car :
Pour toutes ces raisons, on ne peut se contenter des résultats obtenus chez l’adulte.
Il est donc nécessaire, pour une meilleure efficacité et sécurité, d’évaluer chez les enfants et les adolescents les médicaments et les dispositifs médicaux qui leur sont destinés.
Les enfants et adolescents sont particulièrement bien protégés par la règlementation.
Chez l’enfant et l’adolescent, les essais ne peuvent être menés que s’il est impossible d’effectuer des recherches d’une efficacité comparable sur des personnes majeures, et si un bénéfice potentiel, pour l’enfant ou pour tous les enfants est attendu. Dans ce cas, les risques prévisibles et les contraintes de la recherche doivent avoir un caractère minimal.
Les enfants et adolescents ne peuvent participer à un essai sans le consentement de leurs parents ou de leur représentant légal, ni sans leur avis s’ils sont en âge de le donner. Ils doivent recevoir une information adaptée à leur âge. Un refus de participer de l’enfant ou de l’adolescent doit être respecté.
Si l’enfant ou l’adolescent devient majeur pendant la durée de l’essai, son consentement doit à nouveau lui être demandé.
Les comités de protection des personnes (CPP) vérifient que les conditions de protection sont bien respectées avant de donner leur approbation à la réalisation d’un essai chez l’enfant.
Pour aller plus loin, écoutez le podcast du Prof. Régis Hankard qui nous explique pourquoi la recherche clinique en pédiatrie est indispensable.
En France, la mise en œuvre des essais cliniques est réglementée par le code de la santé publique dont une partie est spécifiquement dédiée aux recherches qui impliquent la personne humaine.
La partie législative (loi) est complétée d’une partie réglementaire (décret et arrêtés) donnant plus de précisions sur les aspects opérationnels.
Des bonnes pratiques cliniques (texte réglementaire ou normatif) définissent les rôles et responsabilités des promoteurs et des investigateurs ainsi que tous les principes qui régissent le déroulement des essais cliniques.
Pour les recherches portant sur des médicaments et pour les recherches portant sur des dispositifs médicaux, la règlementation est identique dans tous les pays européens.
L’ensemble des textes qui régissent les essais cliniques de médicaments et de dispositifs médicaux est disponible sur le site de l’agence nationale de sécurité du médicament et des produits de santé (ANSM, www.ansm.sante.fr) :
Avant de démarrer un essai clinique, il est indispensable d’obtenir un avis favorable d’un comité de protection des personnes (CPP) et en fonction de la catégorie de la recherche, une autorisation de l’agence nationale de sécurité des médicaments et des produits de santé (ANSM).
Le CPP doit rendre un avis sur les conditions de validité de la recherche, notamment au regard de sa pertinence.
Un essai clinique qui ne serait pas basé sur le dernier état des connaissances scientifiques et qui ne serait pas nécessaire pour étendre ces connaissances ne pourrait donc pas se dérouler.
ℹ️ Nombre d’essais autorisés par l’ANSM & Infographie du Leem sur la place de la France dans les essais cliniques
Pour la catégorie de recherches cliniques qui comportent une intervention sur la personne non justifiée par sa prise en charge habituelle, le nombre d’essais autorisés par l’ANSM en 2024 est le suivant :

Source : Rapport d’activité ANSM 2024 [PDF]
Infographies Leem, Mars 2025

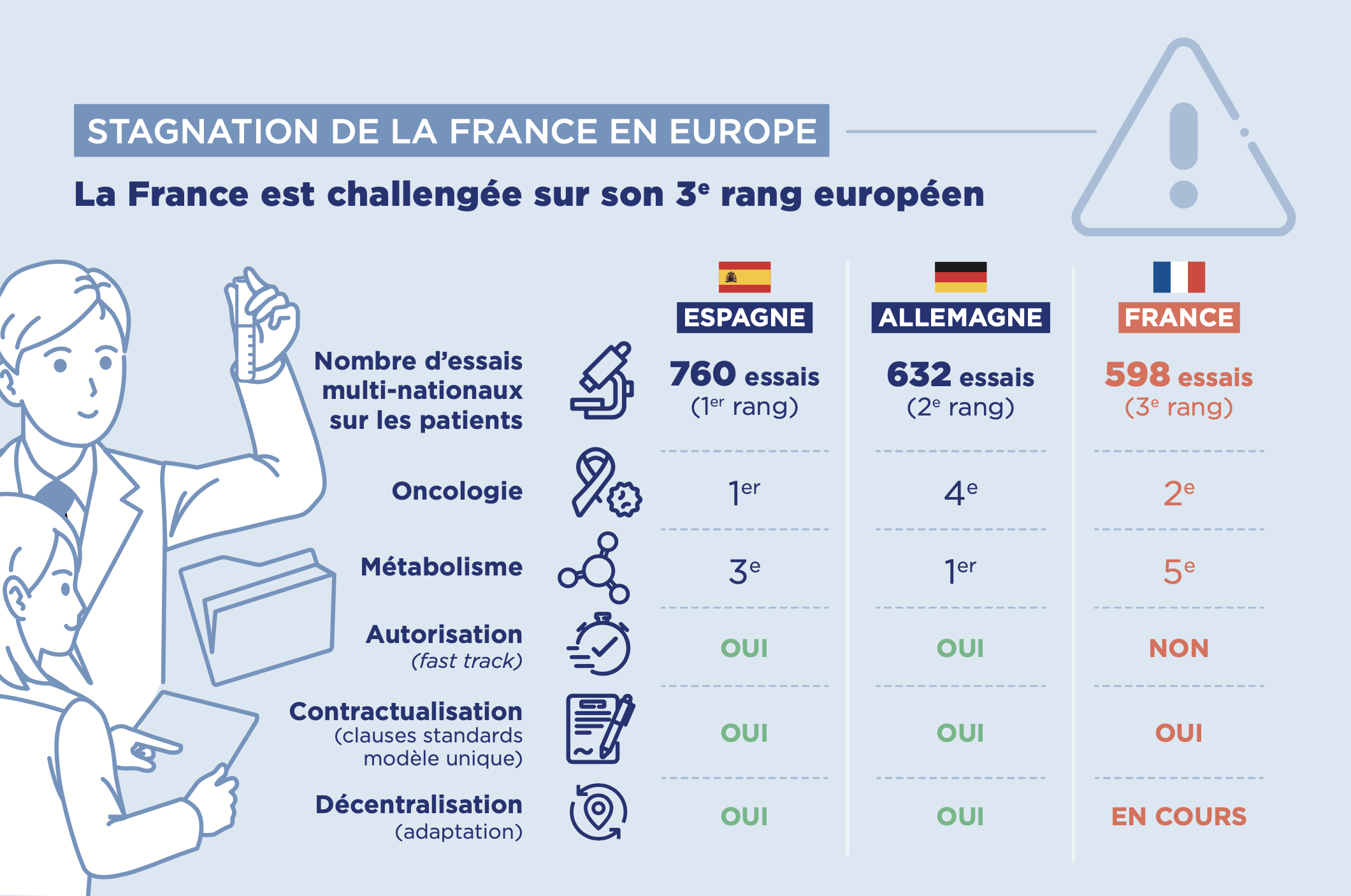
Source : « Recherche clinique : la France stagne, l’Europe recule – Comment préserver l’innovation ? » 18 mars 2025
La recherche et le développement de médicaments et de dispositifs médicaux est réalisée dans le monde entier. Environ 87000 recherches cliniques sont en cours de recrutement dans le monde (recherches interventionnelles et recherches observationnelles). Près de 9% de ces recherches sont conduites en France [Source : ClinicalTrials.gov].
La France est un des principaux pays européens en nombre de recherches réalisées.
Dans le secteur de la santé, la France propose un environnement adéquat pour mener des essais cliniques :
Les principaux domaines de recherche clinique dans lesquels la France est impliquée, sont :
Pour les médicaments :
Pour les dispositifs médicaux :
Cette place privilégiée que la France occupe en recherche clinique est le fruit de la volonté et de l’énergie déployée par tous les acteurs de la recherche clinique, que ce soient les autorités de santé, les établissements de santé, les promoteurs, les investigateurs et bien sûr les patients afin qu’ils aient accès à des thérapies innovantes.
Votre participation permet d’améliorer le traitement, le diagnostic et la prévention des pathologies.
D’un point de vue individuel, participer à un essai clinique vous permet de bénéficier, dans les meilleures conditions de sécurité possibles, à un stade précoce, d’un traitement ou d’un dispositif médical innovant qui n’est pas encore disponible sur le marché.
Pour les volontaires sains (qui ne sont pas malades), participer aux essais permet de s’impliquer dans une démarche d’intérêt général en contribuant au progrès médical.
Dans tous les cas, l’investigateur s’engage à vous délivrer les meilleurs soins possibles.
A priori, tout le monde peut participer à un essai clinique : hommes, femmes, enfants, personnes âgées.
Il est indispensable d’être affilié à un régime de sécurité sociale ou bénéficiaire d’un tel régime.
Une attention particulière est portée aux personnes vulnérables (femmes enceintes, parturientes, mères qui allaitent, personnes privées de liberté par une décision judiciaire ou administrative, personnes faisant l’objet de soins psychiatriques, personnes admises dans un établissement sanitaire ou social à d’autres fins que la recherche, personnes majeures faisant l’objet d’une mesure de protection légale (tutelle, curatelle par exemple) ou hors d’état d’exprimer leur consentement, patients mineurs). Ces personnes peuvent participer si l’importance du bénéfice attendu pour elles-mêmes est de nature à justifier le risque ou si ces recherches se justifient au regard du bénéfice attendu pour d’autres personnes se trouvant dans la même situation. Dans ce cas, les risques prévisibles et les contraintes de la recherche doivent présenter un caractère minimal.
Pour un essai clinique en particulier, il est normal de retenir la population concernée par la pathologie à laquelle va s’adresser le médicament ou le dispositif médical.
Pour pouvoir participer à un essai, la personne devra répondre à certains critères (critères d’éligibilité) très précis qui permettront de répondre à la question posée dans l’essai.
Ces critères peuvent être des critères indispensables pour pouvoir inclure le patient dans l’essai (critères d’inclusion), comme par exemple la présence de la pathologie, certains paramètres biologiques, l’âge, le sexe, l’état général du patient. D’autres critères, s’ils sont présents, empêchent la participation de la personne (critères d’exclusion), comme par exemple la coexistence d’une autre maladie grave ou la prise de certains médicaments.
Il est tout à fait possible d’inclure des femmes dans les essais cliniques, une vigilance particulière sera portée aux moyens de contraception utilisés, afin d’éviter la survenue d’une grossesse au cours de l’essai.
De même, des enfants peuvent être inclus (avec l’autorisation de leurs parents) dans des essais cliniques pédiatriques adaptés à leur âge et à leur pathologie.
Le consentement est l’accord de la personne qui décide librement de participer à un essai clinique après avoir reçu des explications claires et précises.
Le principe général est l’obligation de donner une information claire et précise aux patients pour toutes les recherches. Les modalités de recueil du consentement peuvent être différentes selon les types d’essais (consentement écrit, oral, simple non-opposition).
| Recherches interventionnelles (intervention sur la personne non justifiée par sa prise en charge habituelle) | Recherches interventionnelles qui ne comportent que des risques et des contraintes minimes pour la personne | Recherches non interventionnelles qui ne comportent aucun risque ni contrainte, dans lesquelles le soin et le traitement sont habituels
|
Recherches sur les dispositifs médicaux (investigations cliniques) | |
| Type de consentement | Écrit (document signé et daté) | Demandé oralement, documenté dans le dossier médical du patient | Demande de la non opposition du patient à l’utilisation de ses données, documentée dans le dossier médical du patient
|
Écrit (document signé et daté) |
Dans le cadre des recherches interventionnelles et des investigations cliniques, l’investigateur doit vous expliquer l’essai auquel il vous propose de participer, ses modalités pratiques de déroulement, les alternatives médicales, les bénéfices attendus, les éventuelles contraintes et effets indésirables possibles. Ses explications sont complétées par un document écrit qui vous est remis : le document d’information.
Ce document regroupe toutes les informations nécessaires à la compréhension de l’essai afin de vous permettre de décider d’y participer ou non. Il vous sera demandé de le lire attentivement, de poser toutes les questions que vous avez à l’investigateur en charge de l’essai et de prendre éventuellement un temps de réflexion avant de vous décider. Vous êtes libre de participer ou non à l’essai.
Le document de consentement est la preuve écrite que vous avez lu la note d’information qui vous a été donnée et que vous acceptez de participer à l’essai. Il doit être impérativement signé avant le début de l’essai.
L’absence de signature interdit votre participation à l’essai.
Votre signature ne décharge par l’investigateur de son devoir de vous apporter les meilleurs soins possibles.
Le consentement peut être retiré à tout moment par le patient qui le souhaite, sans justification, il faut cependant en informer l’investigateur de la recherche afin qu’une prise en charge médicale adaptée soit effectuée.
Participer à un essai clinique ne coûte rien, mais cela ne peut pas être considéré comme un travail donnant lieu à une rémunération.
Les frais éventuels engagés (par exemple les frais de transport ou de stationnement, les frais de restauration) sont remboursés par le promoteur en totalité sur justificatif. Cela doit être expliqué par l’investigateur en même temps que les informations sur l’essai.
De façon exceptionnelle, dans certains essais, comme par exemple des essais sur des personnes non malades, des essais nécessitant une immobilisation ou un temps de présence très long, le patient peut être indemnisé en compensation de contraintes subies. Le comité de protection des personnes (CPP) se prononce sur les montants et les modalités d’indemnisation des participants. Le montant total de ces indemnités est fixé par un texte officiel, il ne peut excéder 6000 euros par période de 12 mois consécutifs.
Sous réserve de l’acceptation par le comité de protection des personnes, le versement des indemnités aux volontaires sains peut être effectué par des avantages en nature, lorsque deux conditions sont remplies :
– le promoteur a fait une évaluation financière de ces avantages en nature et vérifié que le montant fixé (6000 euros par période de 12 mois consécutifs) n’est pas dépassé ;
– le promoteur a justifié le recours à ces avantages en nature au regard des spécificités de la recherche ou de la population visée.
De plus, les indemnités sont renseignées par l’investigateur sur le fichier national des personnes qui se prêtent à des recherches, afin de s’assurer du respect du montant maximum autorisé. Ce fichier est géré par le Ministère de la santé.
Le versement d’une telle indemnité est interdit dans le cas des recherches effectuées sur des mineurs, des majeurs protégés (par exemple sous tutelle ou sous curatelle) ou hors d’état d’exprimer leur consentement, des personnes privées de liberté, des personnes hospitalisées sans leur consentement ou admises dans un établissement sanitaire ou social à d’autres fins que la recherche.
Les participants à un essai ont des droits qu’ils doivent connaître.
Ces droits sont les suivants :
Il ne faut pas hésiter à poser des questions à l’investigateur dès qu’une interrogation survient sur l’un de ces points.
Replay du webinaire Notre Recherche Clinique qui a eu lieu le 27 mai 2025, à l’occasion de la journée internationale de la recherche clinique, un webinaire gratuit et ouvert à toutes et tous, pour répondre à toutes vos questions sur la recherche clinique.
3 experts étaient mobilisés pour cet événement :
Dr. Soizic Courcier, expert recherche clinique et affaires médicales
Prof. Dominique Deplanque, médecin neurologue, coordonnateur du Centre d’Investigation Cliniques 1403 INSERM du CHU de Lille. Dominique Deplanque est également Président de la Société Française de Pharmacologie et de Thérapeutique, il a contribué à nos podcasts ‘Idées reçues sur les essais cliniques‘
Christophe Demonfaucon, membre de l’AFTOC, l’association française des personnes souffrant de troubles obsessionnels compulsifs, membre d’un Comité de Protection de Personnes et représentant de France Asso Santé auprès du comité éditorial Notre Recherche Clinique
Animation par Claire Sibenaler de l’équipe éditoriale Notre Recherche Clinique.